[1]:
import scanpy as sc
import pandas as pd
from anndata import AnnData
from pathlib import Path
import json
from matplotlib.image import imread
from step import scModel, stModel
from step.utils.misc import read_visium_hd
sc.set_figure_params(dpi=150, figsize=(6, 4.5))
/projects/82505004-e7a0-445f-ab3c-80d03c91438f/.cache/pypoetry/virtualenvs/step-Ajq_Bw_i-py3.10/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Visium HD Human Colorectal Cancer (16 um) cell type clustering & spatial domain identification¶
16 um bin size: cell type clustering¶
[ ]:
adata = read_visium_hd("./data/visium-hd/human-coloretal-cancer/square_016um/")
[3]:
stepc = scModel(
adata=adata,
n_top_genes=2000,
)
Trying seurat_v3 for hvgs
not log_transformed
Adding count data to layer 'counts'
================Dataset Info================
Batch key: None
Class key: None
Number of Batches: 1
Number of Classes: None
Gene Expr: (136984, 2000)
============================================
[4]:
stepc.run(epochs=400, batch_size=2048, beta=1e-3)
Performing global random split
Current Mode: single_batch: ['gene_expr']
50%|█████ | 202/400 [18:05<17:37, 5.34s/epoch, kl_loss=0.956, recon_loss=460.729, val_kl_loss=0.967/0.498, val_recon_loss=458.524/457.657]Early Stopping triggered
50%|█████ | 202/400 [18:05<17:43, 5.37s/epoch, kl_loss=0.956, recon_loss=460.729, val_kl_loss=0.967/0.498, val_recon_loss=458.524/457.657]
EarlyStopping counter: 30 out of 30
EarlyStopping counter: 202 out of 10
[5]:
adata = stepc.adata
sc.pp.neighbors(adata, use_rep='X_rep', n_neighbors=60)
sc.tl.umap(adata)
[6]:
sc.tl.leiden(adata)
sc.pl.umap(adata, color='leiden')
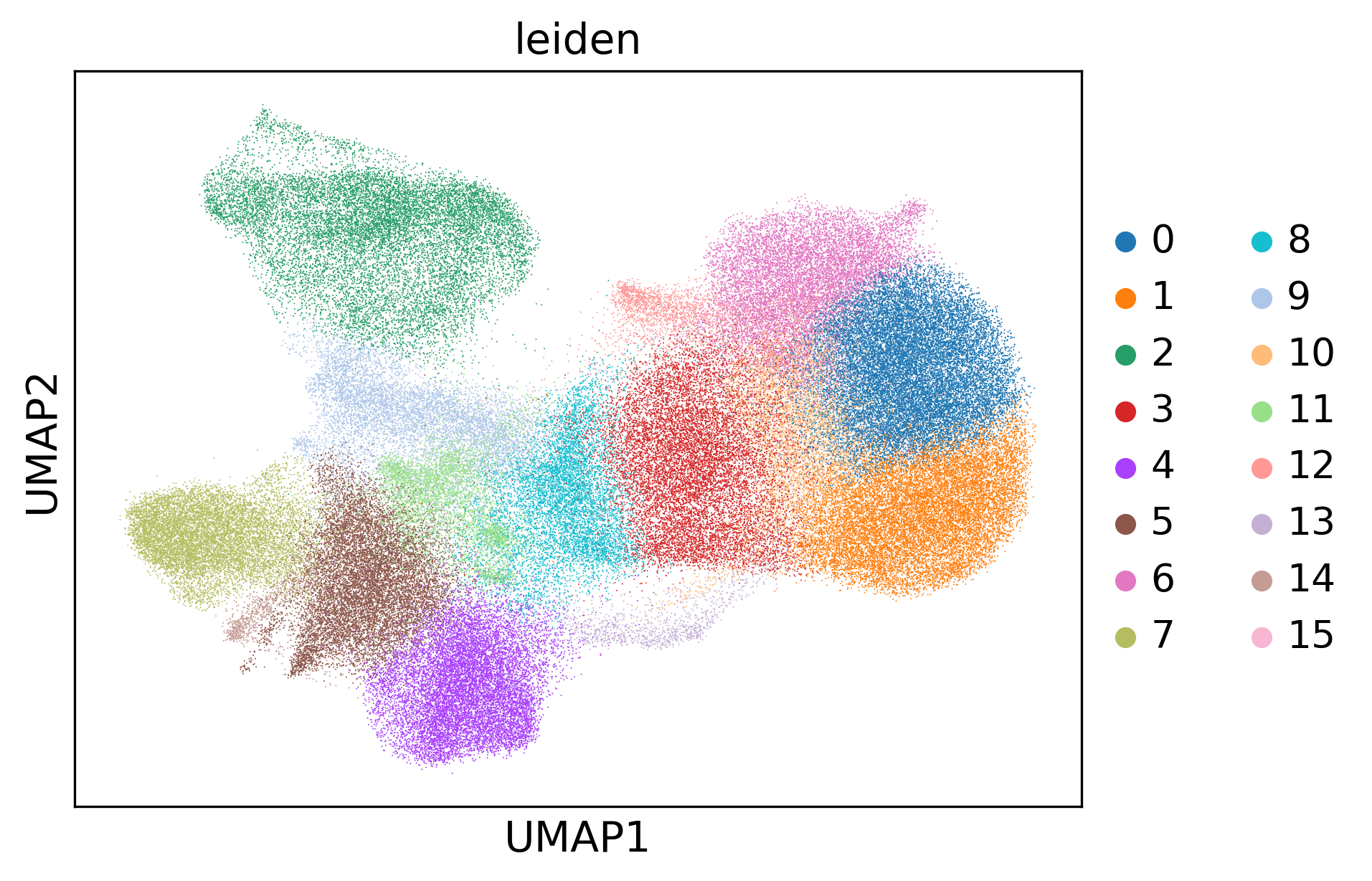
[7]:
sc.pl.spatial(adata, color='leiden')

[8]:
stepc.save("./results/visium-hd/hcc-16um/")
adata.write_h5ad("./results/visium-hd/hcc_16um.h5ad")
Saving model...
Saving model config...
Saving dataset config...
[2]:
adata = sc.read_h5ad("./results/visium-hd/hcc_16um.h5ad")
# sc.set_figure_params(dpi=300, dpi_save=300)
# sc.settings.figdir = './results/visium-hd/hcc-16um/'
[6]:
sc.pl.spatial(adata, color='leiden', frameon=False, groups=['7', '11', '14', '2', '9', '10'], na_in_legend=False)

[3]:
sc.settings.figdir = "./results/visium-hd/hcc-16um/"
sc.set_figure_params(dpi_save=300)
[4]:
sc.tl.rank_genes_groups(adata, groupby='leiden', dendrogram=False, use_raw=True)
sc.tl.dendrogram(adata, use_raw=True, groupby='leiden')
WARNING: You’re trying to run this on 2000 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
[5]:
sc.pl.rank_genes_groups_matrixplot(adata,
groupby='leiden',
values_to_plot='logfoldchanges',
cmap='RdBu_r',
vmin=-4, vmax=4,
save='16um_leiden.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/matrixplot_16um_leiden.svg
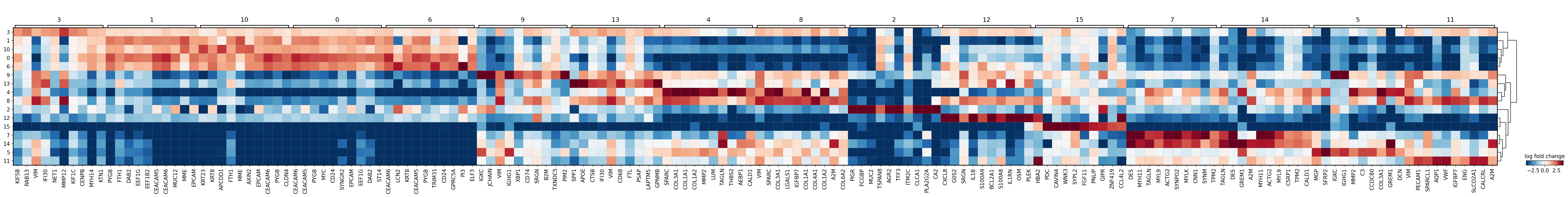
[9]:
sc.pl.rank_genes_groups_stacked_violin(adata,
groupby='leiden',
save='_16um_leiden.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/stacked_violin__16um_leiden.svg
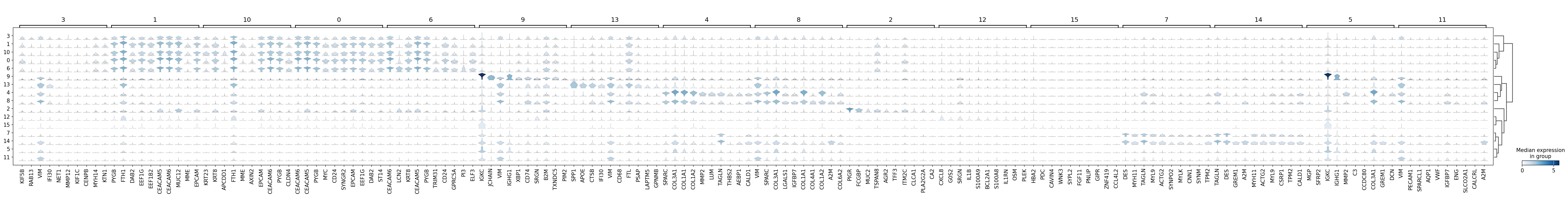
[12]:
sc.pl.rank_genes_groups_heatmap(adata,
groupby='leiden',
n_genes=10,
swap_axes=True,
vmax=4,
show_gene_labels=True,
save='_16um_leiden.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/heatmap_16um_leiden.svg
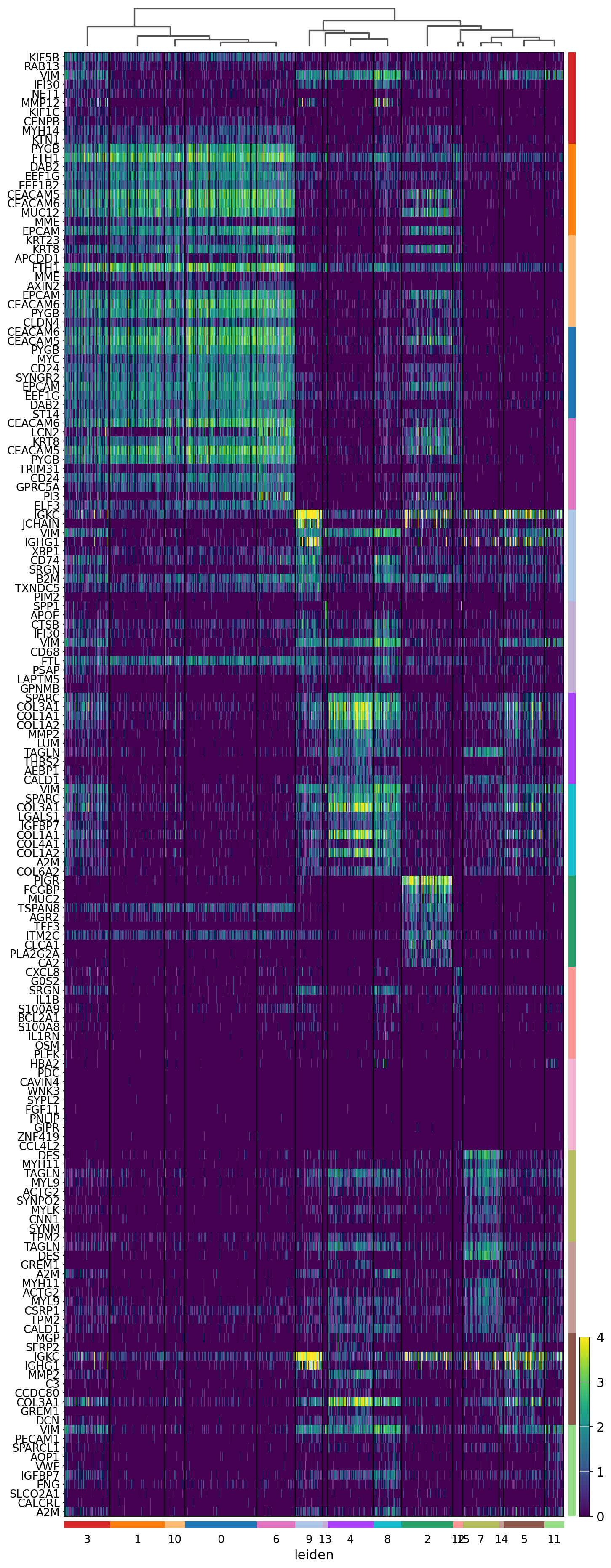
16 um bin size: spatial domain identification¶
[2]:
adata = read_visium_hd("./data/visium-hd/human-coloretal-cancer/square_016um/")
[3]:
stepc = stModel(
adata=adata,
n_top_genes=3000,
edge_clip=2,
n_glayers=3,
)
Trying seurat_v3 for hvgs
not log_transformed
Adding count data to layer 'counts'
Dataset Done
================Dataset Info================
Batch key: None
Class key: None
Number of Batches: 1
Number of Classes: None
Gene Expr: (136984, 3000)
============================================
[4]:
stepc.run(n_iterations=4000, n_samples=2048, beta=1e-5)
Training with e2e pattern
Training graph with single batch
Constructing graph for batch
100%|██████████| 4000/4000 [06:14<00:00, 10.67step/s, recon_loss=1340.246, kl_loss=0.002, contrast_loss=1.701, graph_ids=None]
[5]:
stepc.cluster(n_clusters=12)
stepc.spatial_plot(color='domain',)
stepc.save("./results/visium-hd/hccspatial_plotomain/")
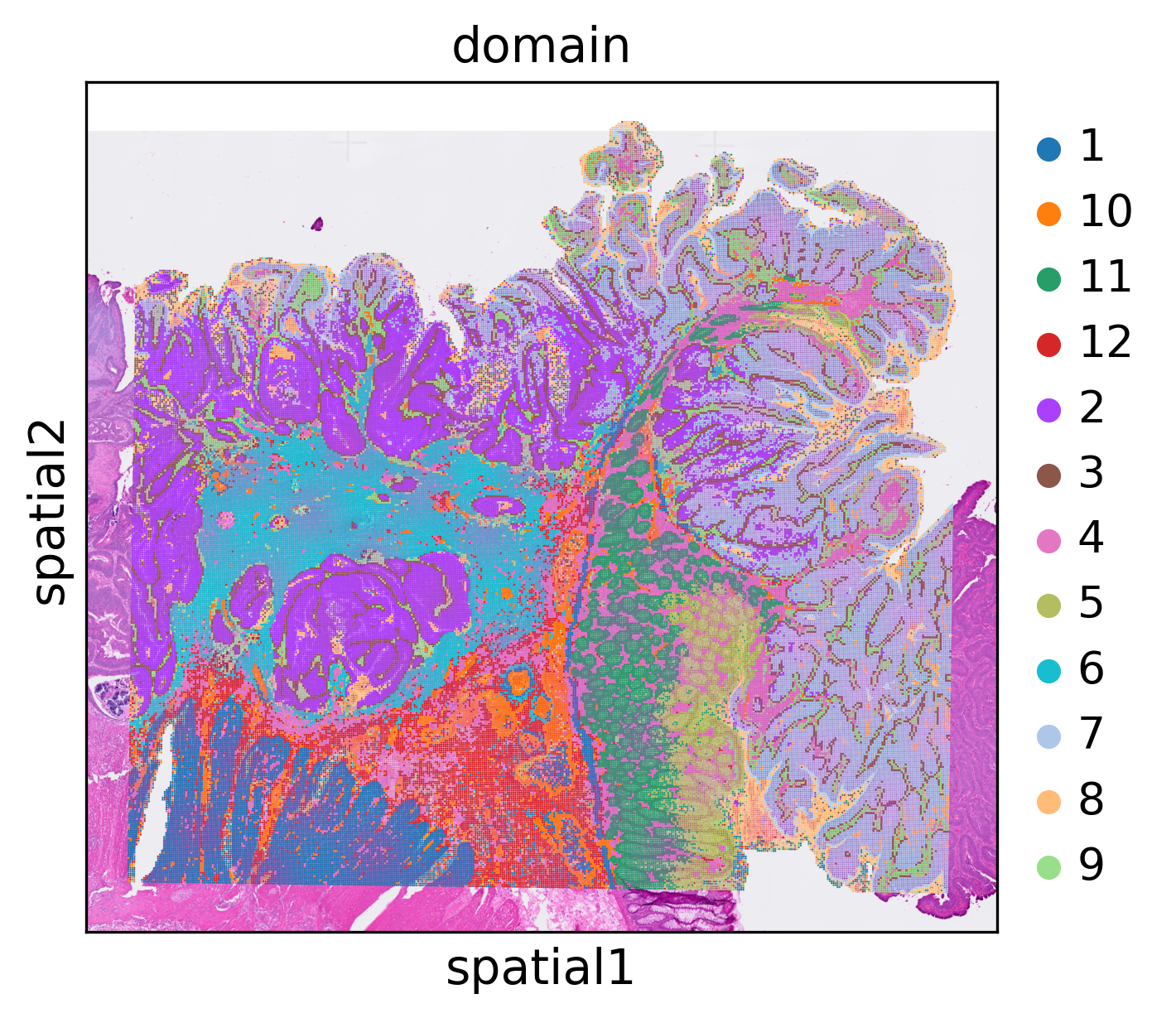
[8]:
stepc = stModel.load("./results/visium-hd/hccspatial_plotomain/",
filepath="./results/visium-hd/hcc_16um_domain.h5ad")
stepc.adata.obsm['X_smoothed'] = stepc.gembed()
stepc.cluster(n_clusters=8)
stepc.spatial_plot(color='domain',)
Trying seurat_v3 for hvgs
Adding count data to layer 'counts'
Dataset Done
================Dataset Info================
Batch key: None
Class key: None
Number of Batches: 1
Number of Classes: None
Gene Expr: (136984, 3000)
============================================
Loading backbone model...
Backbone model loaded.
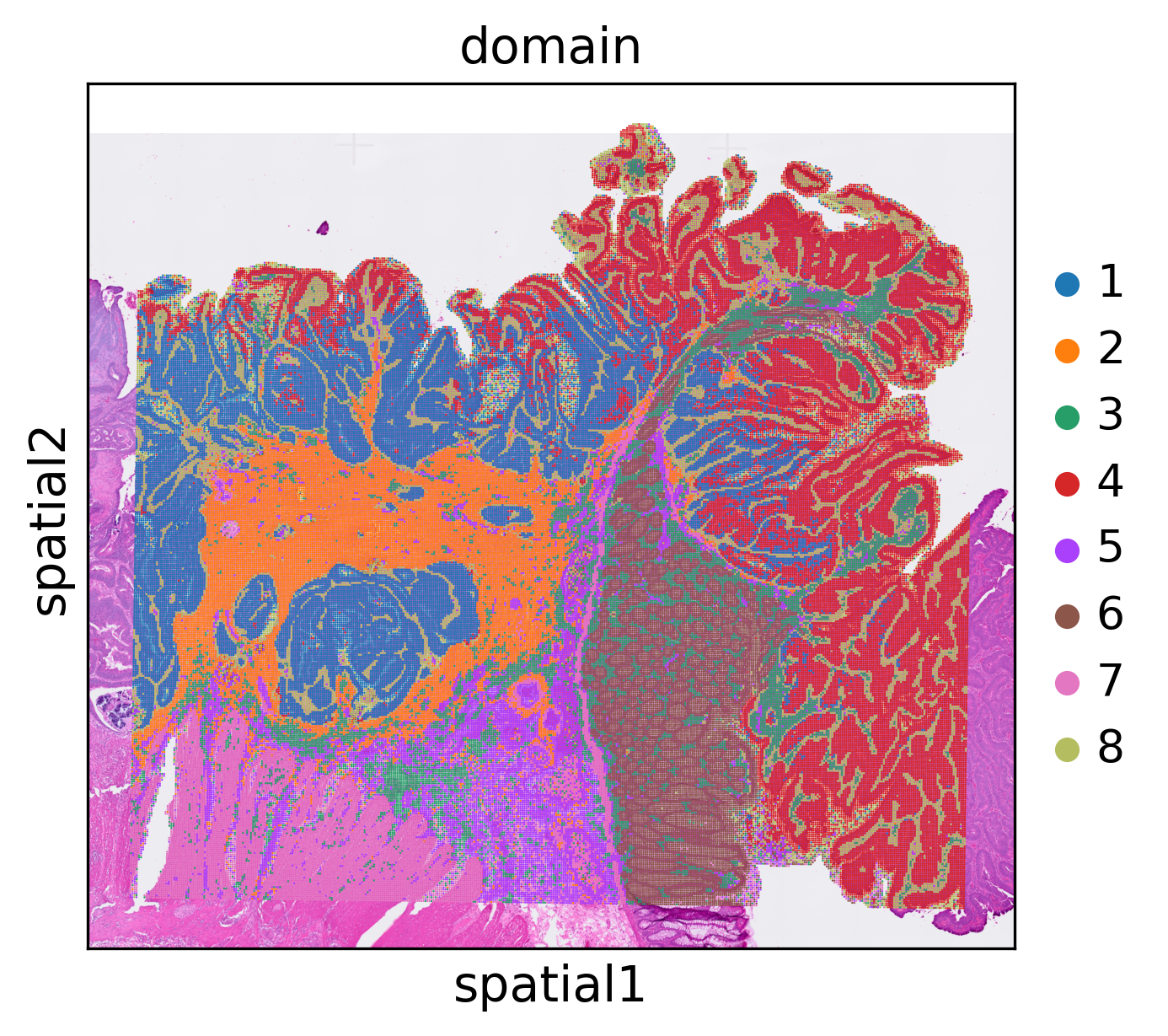
[9]:
stepc.adata.write_h5ad("./results/visium-hd/hcc_16um_domain.h5ad")
[2]:
adata = sc.read_h5ad("./results/visium-hd/hcc_16um_domain.h5ad")
[3]:
sc.settings.figdir = "./results/visium-hd/hcc-16um/"
sc.set_figure_params(dpi_save=300, figsize=(12, 9))
sc.pl.spatial(adata, color='domain', frameon=False, show=False, save='_16um_domain.svg',)
[4]:
adata_raw = adata.raw.to_adata()
sc.pp.log1p(adata_raw)
adata.raw = adata_raw
[5]:
# adata = stepc.adata
sc.tl.rank_genes_groups(adata, groupby='domain', dendrogram=False, use_raw=True)
sc.tl.dendrogram(adata, use_raw=True, groupby='domain')
WARNING: You’re trying to run this on 3000 dimensions of `.X`, if you really want this, set `use_rep='X'`.
Falling back to preprocessing with `sc.pp.pca` and default params.
[22]:
sc.pl.rank_genes_groups_matrixplot(adata,
groupby='domain',
values_to_plot='logfoldchanges',
cmap='RdBu_r',
vmin=-4, vmax=4,
save='16um_domain.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/matrixplot__16um_domain.svg
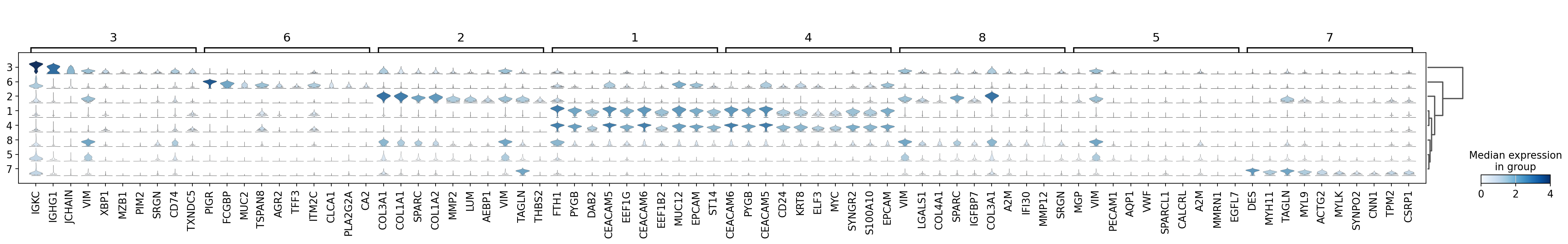
[7]:
sc.pl.rank_genes_groups_stacked_violin(adata,
groupby='domain',
vmax=4,
save='16um_domain.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/stacked_violin_16um_domain.svg
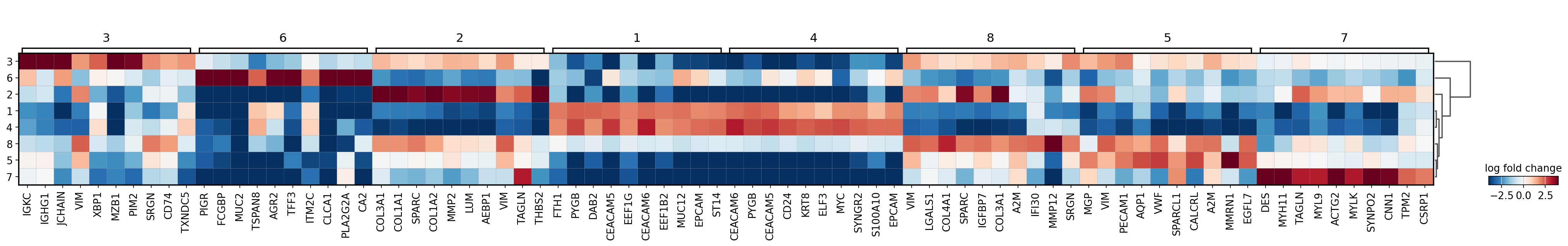
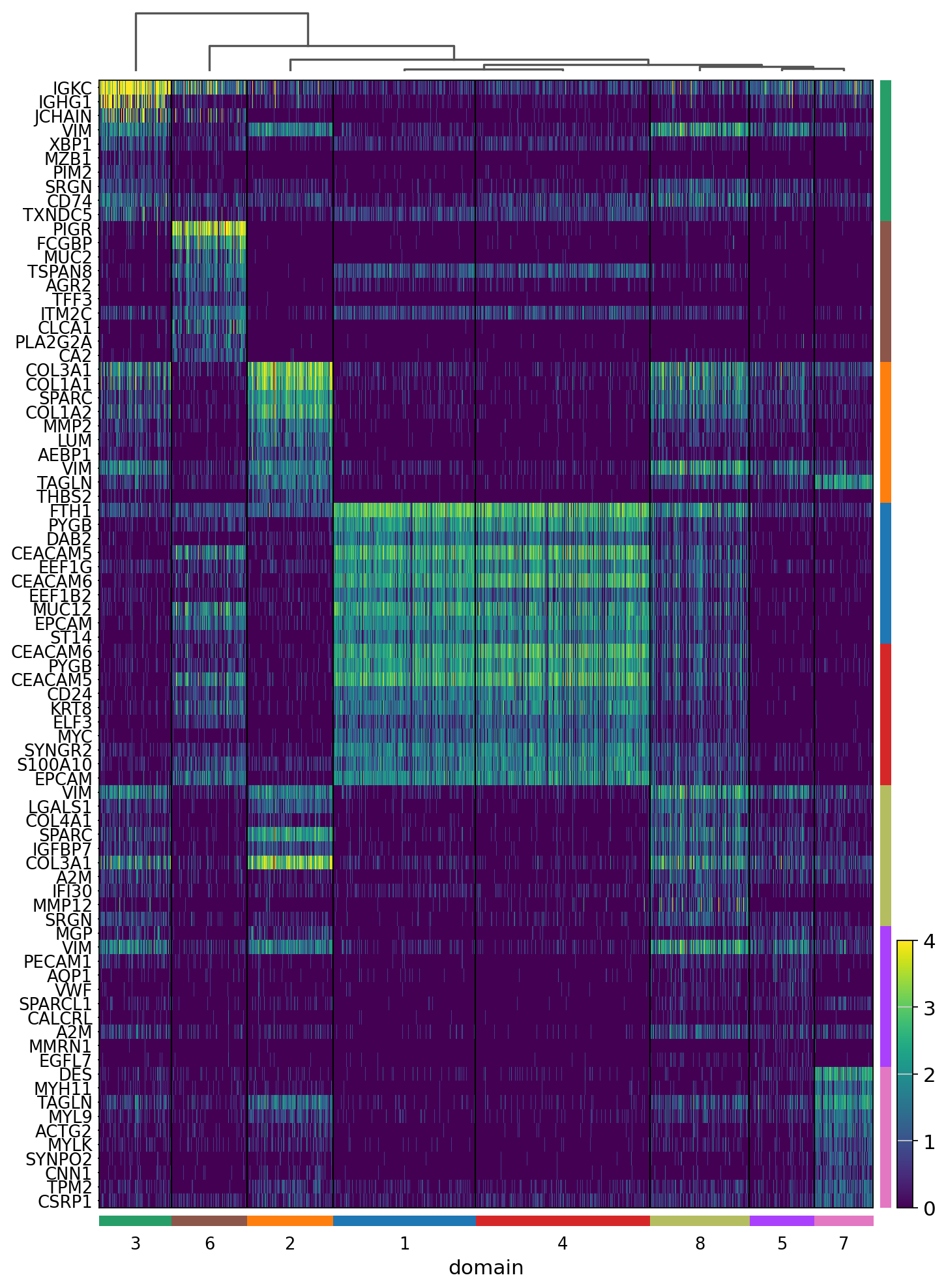
[23]:
sc.pl.rank_genes_groups_heatmap(adata,
groupby='domain',
n_genes=10,
swap_axes=True,
vmax=4,
show_gene_labels=True,
save='_16um_domain.svg')
WARNING: saving figure to file results/visium-hd/hcc-16um/heatmap_16um_domain.svg
[ ]: